Amyloid aggregation: functional and disease related
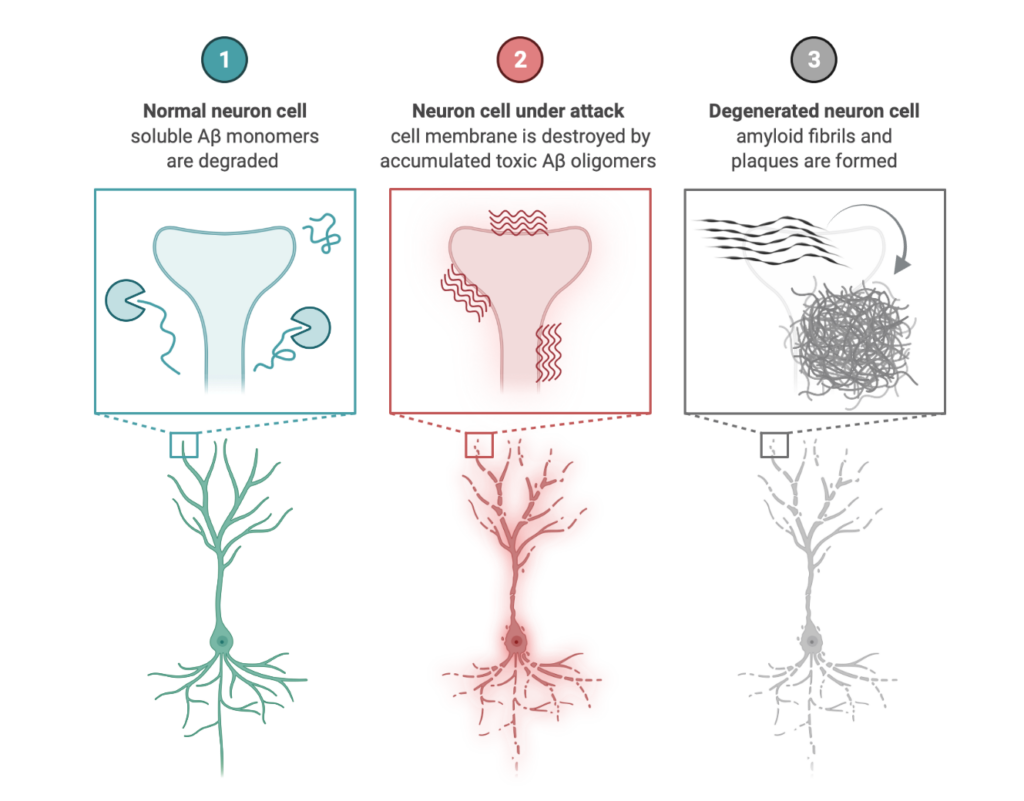
Amyloid protein aggregation is a wide-ranging phenomenon and is of great importance in several areas of science as such aggregates can lead to diseases or functional and contributing to normal biological processes.Unfortunately, the detailed molecular mechanism of the amyloid aggregation is still not fully resolved. To this end, we are employing full-atomistic or coarse-grained molecular dynamics simulations of amyloid-beta peptide (Aβ), human islet amyloid polypeptide (hIAPP), Huntingtin, PI3-Kinase at the presence of neuronal membranes, carbohydrates, molecular crowders and/or at different ionic strengths to gain insight on the possible biophysical mechanisms of amyloid aggregation manifested through the following research project:
Simulating Aβ-membrane interactions considering in vivo conditions.
Developing hybrid simulation models to study Aβ42 aggregation.
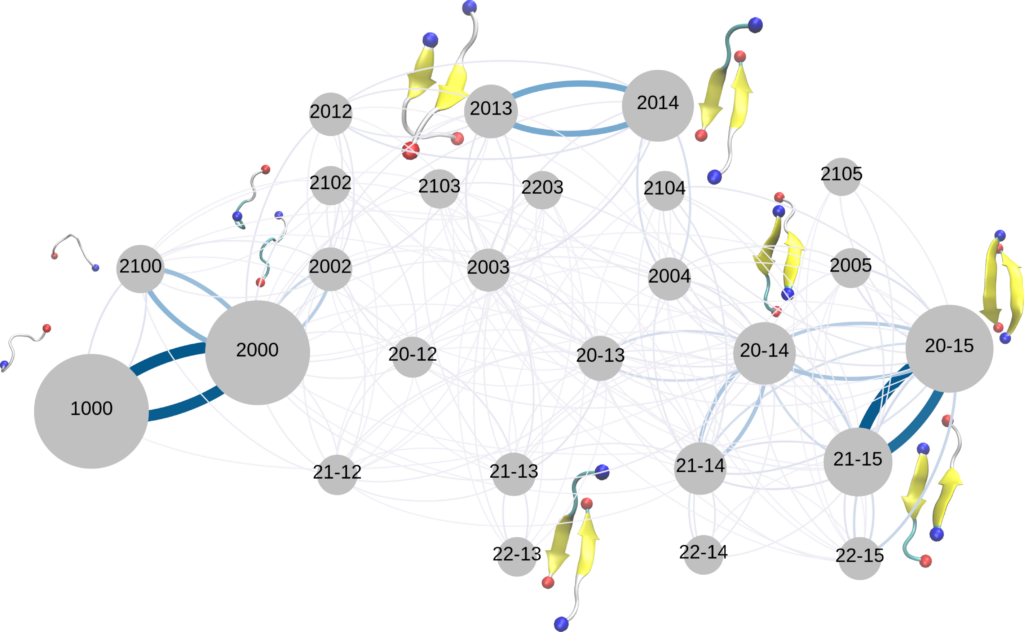
Simulating Amyloid aggregation in solution.
Exploring intrinsically disordered proteins and their propensity to aggregate into amyloid beta structures.ı
Pathogen-host interactions provoke the immune reaction of γ-interferons, which in turn regulate a multitude of proteins. Upon Toxoplasma gondii infection, murine guanylate binding proteins (mGBPs) prove vital for mice survival. Here, mGBPs localize to the parasitophorous vacoule membrane of the parasite. In order to elucidate their mechanism of action, we perform atomistic and coarse-grained MD simulations, generating results about binding mode, lipid specificity and possible membrane disruption. Furthermore, we study the dynamics of a dynamin-like hinge motion using enhanced sampling methods and possible dimer structures using docking. Close collaboration with experimentators from CRC1208 (HHU, Klaus Pfeffers group, Institute of Medical Microbiology and Hospital Hygiene) allows continuous cross-verification of our joined findings.
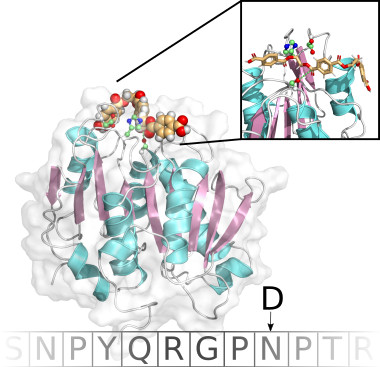
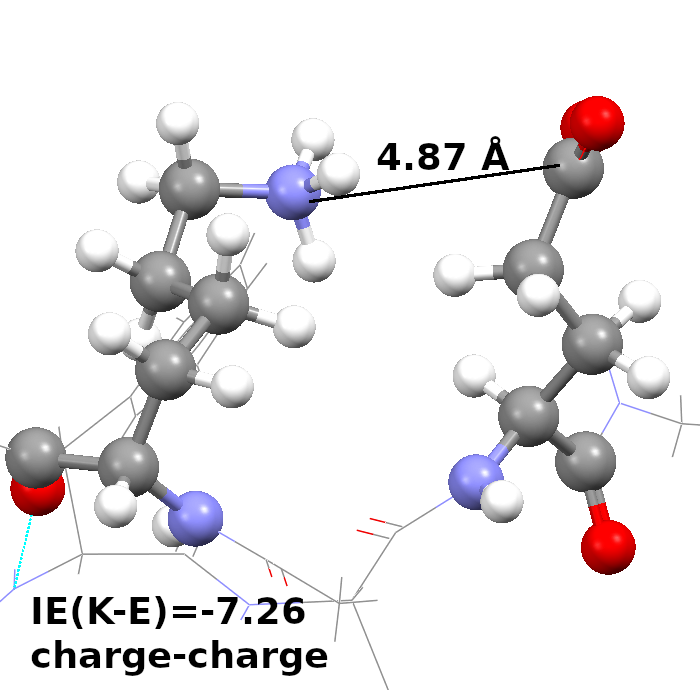
Sickle cell disease (SCD) is an inherited blood disorder resulting from a single point mutation in the beta-globin gene involving the substitution of glutamic acid at the sixth position of the β-globin chain of the hemoglobin molecule to valine (E6V). Under low oxygen tension, the highly hydrophobic patch formed by the E6V substitution results in HbS aggregation due to the attraction of the mutated hydrophobic valine to a hydrophobic pocket of an adjacent HbS molecule, formed by several hydrophobic residues, namely; Ala-β70, Phe-β85, and Leu-β88. Understanding the structural changes that characterize the aggregation path of HbS is very critical in identifying novel molecules that are capable of inhibiting HbS aggregation. Using Molecular Dynamics Simulations, we study the aggregation of HbS and identify aggregation hotspots. We also employ Computer Aided Drug Design methods to identify new molecules that are capable of binding to the aggregation prone-sites and thereby inhibit aggregation.
This project is funded by Forschungszentrum Jülich.
Computational enzyme design
Enzymatic biocatalysis constitutes a promising environmentally friendly alternative to harmful industrial processes, but the stability, activity and selectivity/specificity is most often not sufficient for biotechnological application.In collaboration with experimental groups, as well as experts in highly contemporary computational methods, we address the engineering of organic solvent tolerant esterases, poly(ethylene) terephthalate (PET) degrading enzymes and cytochrom P450 BM3 using all-atom standard and accelerated molecular dynamics (MD) as well as hybrid quantum mechanics/molecular mechanics (QM/MM) simulations complemented by state-of-the-art machine learning for structure-activity relationship predictions.
This project is funded by Deutsche Bundesstiftung Umwelt.
Computer-aided drug design
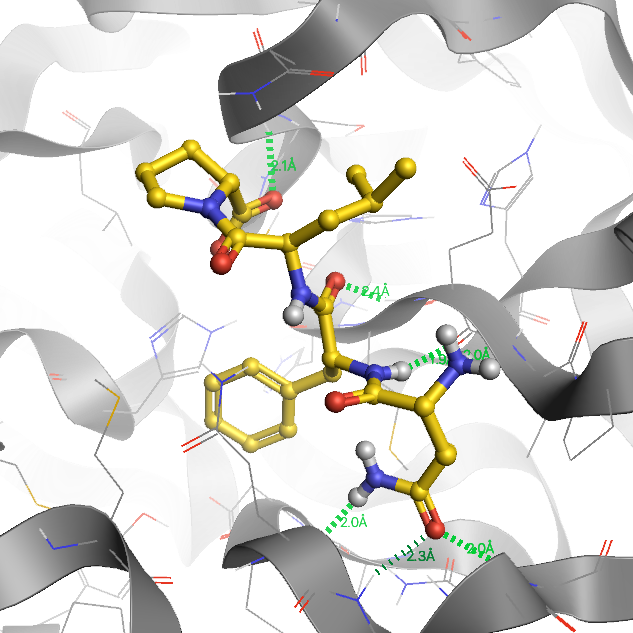
When COVID-19 became a global pandemic in the spring of 2020, we initiated the computer-aided drug design (CADD) project to contribute to the discovery and development of a prospective drug against SARS-CoV-2. We screened over one million compounds by docking them against the main protease (3CLpro) and used molecular dynamics (MD) simulations to identify compounds with the highest binding affinity. In our current work, we aim to test D-peptides for their inhibitory activity against 3CLpro and use CADD to improve the pharmacokinetics and drug efficacy of the best candidates. Together with our collaborators at the Forschungszentrum Jülich and the Heinrich-Heine University we are able to investigate the ligands holistically for their drug potential through in silico, in vitro and in cell approaches.
Force fields are at the heart of every simulation and their effects on the outcome depend very sensitively on their quality. Force fields that are taking the intrinsically disordered nature of the proteins into account only started to emerge in the last couple of years. However, their performance still depends mostly on the studied system as it has been recently shown that the choice of a force field has a larger impact on the simulation outcomes than the studied protein sequences. As a result, a systematic review and verification of the force fields are very important. We are combining all-atom molecular dynamics simulations and quantum mechanical calculations to verify the performance of the force fields as well as providing data and insight to help improving them with the focus on amyloid aggregation.
This project is funded by Forschungszentrum Jülich.
Methods and software development
Hybrid force fields that combine all-atom and coarse-grained representations of the studied systems have been gaining more attention in the recent years to solve the time-scale problem in the molecular dynamics simulation. For instance, representing the protein using an all-atom force field while treating the solvent with a coarse-grained model could help reaching longer simulation times, with the cost of accuracy. We are working on developing such a force field for the amyloid-beta 42 protein to further understand its aggregation.
We develop software for the prediction of mutation effects on enzyme properties, including enantioselectivity, absoprtion, and fluorescence. To this end, we investigate the application of machine learning approaches, including transfer learning from large genome databases, to create precise predictions. Furthermore, we are maintaining Python codes to calculate and build transition networks for protein aggregation.
All the software and force fields developed by our group are available publicly on our GitHub page: https://github.com/strodel-group
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping our team to understand which sections of the website you find most interesting and useful.
Strictly Necessary Cookies
Strictly Necessary Cookie should be enabled at all times so that we can save your preferences for cookie settings.
If you disable this cookie, we will not be able to save your preferences. This means that every time you visit this website you will need to enable or disable cookies again.